

Abstract
Introduction: Langerhans cell histiocytosis (LCH) is a clonal neoplastic disorder of CD1a/CD207 positive myeloid dendritic cells with broad clinical spectrum ranging from focal self-limited disease (single system LCH, SS-LCH) to aggressive multisystem disease (multi system LCH, MS-LCH) with increased mortality. Mutually exclusive somatic activating mutations in BRAF(V600E, ~50-65%) and MAP2K1(~25%) genes are reported in LCH patients resulting in overactive MAP kinase pathway. Clinically the disease may affect any organs, more frequently bones, skin, and pituitary gland. Involvement of hematopoietic system, spleen, liver or CNS is considered high risk with less favorable prognosis. Involvement of "special sites" such as craniofacial bones (orbit, mastoid, sphenoid or temporal bones) may pose immediate risk. Systemic therapy is indicated in MS-LCH, SS-LCH with multifocal lesions (e.g. multiple bone lesions) or SS-LCH with special site lesions. Treatment of LCH in adults is challenging due to lack of prospective data and treatment protocols developed in pediatric population is very poorly tolerated in adults. The aim of this study is to review the clinical and genetic characteristics of adult LCH cases presenting to our institute and review treatment paradigm and response.
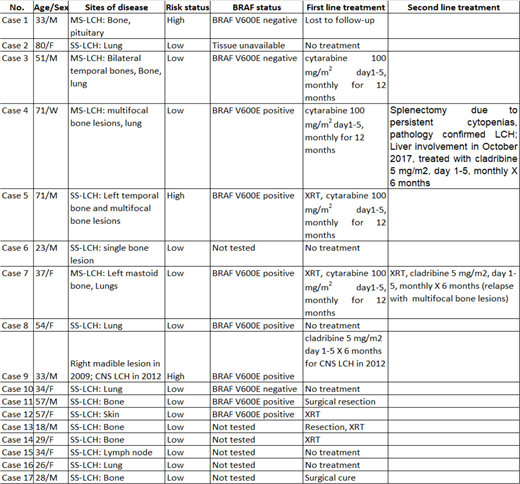
Methods:We retrospectively identified patients reported as "Langerhans cell histiocytosis" between January 2008 to March 2018 by Hematopathology at the ARUP laboratories, University of Utah. Our search identified 23 unique patients; of these 3 cases were excluded due to age <18 years and 3 were excluded due to lack of follow-up records. Seventeen unique adult LCH patients were identified. Data on clinical characteristics, sites of disease involvement, genetic mutations, treatment were obtained by retrospective chart review (Table 1). BRAFV600Emutation analysis was performed by allele specific PCR amplification since 2010 where tissue was available. Detection of the PCR product(s) is performed by capillary electrophoresis and data analysis is done using GeneMarker 2.4.0 software (SoftGenetics; State College, PA, USA).
Results:The median age was 34 years (range 18-80) and 8 patients were men (47%). One patient was initially diagnosed at 3 years of age, recurred at 7 years and 18 years of age and was included. 12 patients had SS-LCH (71%) and 5 patients had MS-LCH (29%). Three (18%) patients were considered high risk at diagnosis (1 liver, and 2 CNS). Bone was the most common site involved in 11 cases (68.7%) followed by lung in 6 cases (37.5%). Of 10 patients tested, 7 (70%) patients were positive for BRAFV600Emutation (Table 1). Six (35.2%) patients had an indication for initiation systemic chemotherapy, one lost to follow-up after presenting with CNS-LCH. 1 patient treated with first line systemic therapy with cladribine 5 mg/m2days 1-5, monthly for 6 months due to CNS-LCH involving hypothalamus. Four received first line therapy with cytarabine 100 mg/m2days 1-5, monthly for 12 months due to multifocal bone lesions or involvement of craniofacial bones; of which two patient recurred (1 patient with recurrent bone lesions and 1 with spleen/liver involvement), cladribine 5 mg/m2days 1-5, monthly for 6 months.
Conclusions: In our single center experience, incidence of adult LCH is extremely rare. Majority of LCH patients presented with SS-LCH. Approximately 30% of the patients required systemic chemotherapy, and recurrence occurred in two patients who required further systemic therapy.Most common indications for systemic therapy are multifocal bone disease or involvement of craniofacial bones or CNS LCH.
Deininger:Blueprint: Consultancy; Pfizer: Consultancy, Membership on an entity's Board of Directors or advisory committees.

